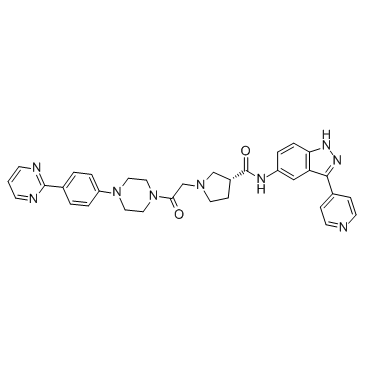
SCH-772984
货号:
IS2200
品牌:
Jinpan
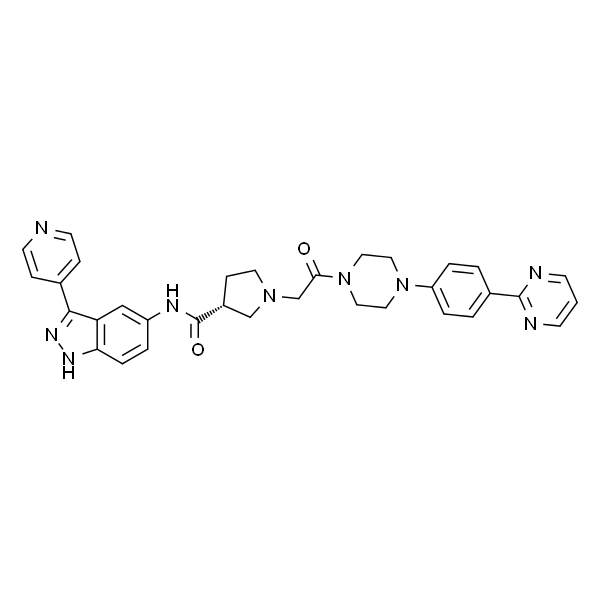
暂无详情
产品简介
| 有效期 | 2年 |
| EC | EINECS 1592732-453-0 |
| 别名 | SCH772984 抑制剂 |
| CAS | 942183-80-4 |
| 分子式 | C33H33N9O2 |
| 分子量 | 587.67 |
| 储存条件 | -20℃ |
| 纯度 | ≥98% |
| 外观(性状) | Solid |
| 单位 | 瓶 |
| 生物活性 | SCH772984 有效抑制 ERK1 和 ERK2 活性,IC50 分别为 4 和 1 nM。[1] |
| IC50 | ERK2:1 nM ; ERK1:4 nM [1] |
| In Vitro | SCH772984分别显示在大约88%和49%的BRAF-突变体(n = 25)或RAS-突变体(n = 35)肿瘤系中EC50值小于500nM。对SCH772984敏感的黑素瘤细胞的流式细胞术分析显示G1停滞以及指示细胞凋亡的亚G1部分的增加。对于RAS和BRAF(n = 61),不到20%的野生型细胞对SCH772984敏感[1]。 |
| In Vivo | 用SCH772984(50mg / kg每天两次)处理BRAF-突变体LOX黑素瘤异种移植物导致98%的肿瘤消退。在KRAS-突变胰腺MiaPaCa模型中也观察到剂量依赖性抗肿瘤活性,在50mg / kg每天两次36%回归。重要的是,肿瘤消退伴随着对肿瘤组织中ERK磷酸化的强烈抑制。通过发病率,致死率或体重减轻来衡量,SCH772984在此时间表上的耐受性良好[1]。 |
| 激酶实验 | 对于纯化的ERK1或ERK2,以8点稀释曲线一式两份地测试SCH772984。将酶加入反应板中并与化合物一起温育,然后加入底物肽和ATP溶液。将14微升稀释的酶(每个反应0.3ng活性ERK2)加入到384孔板的每个孔中。轻轻摇动平板以混合试剂并在室温下温育45分钟。用60μLIMAP结合溶液(在1x结合缓冲液中1:2,200稀释的IMAP珠子)终止反应。将板在室温下再温育0.5小时,以使磷酸肽与IMAP珠完全结合。在LJL Analyst [1]上读取平板。 |
| SMILES | O=C([C@H](CC1)CN1CC(N(CC2)CCN2C(C=C3)=CC=C3C4=NC=CC=N4)=O)NC5=CC6=C(C=C5)NN=C6C7=CC=NC=C7 |
| 靶点 | ERK1,2 |
| 动物实验 | 小鼠[1]裸鼠皮下注射特定细胞系,生长至约100 mm3,随机分为治疗组(10只小鼠/组),腹腔注射SCH772984(12.5,25或50 mg / kg)或载体。肿瘤长度(L),宽度(W)和身高(H)在治疗期间和之后通过卡尺每周两次测量每只小鼠,然后使用公式(L×W×H)/来计算肿瘤体积。 2。每周两次测量动物体重。完成实验后,处理载体和SCH772984处理的肿瘤活组织检查用于Western印迹分析。 |
| 细胞实验 | 细胞增殖实验以96孔格式(六次重复)进行,BRAFV600E-突变型人黑素瘤细胞系LOXIMV1(LOX)以每孔4,000个细胞的密度接种。在细胞接种后24小时,对于所有浓度,细胞用DMSO或9点IC50稀释液(0.001-10μM)处理,终浓度为1%DMSO。使用ViaLight发光试剂盒在给药后5天测定活力。对于细胞系组生存力测定,用SCH772984处理细胞4天,并通过CellTiterGlo发光细胞活力测定法[1]进行测定。 |
| 数据来源文献 | [1]. Morris EJ, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013 Jul;3(7):742-50. |
| 规格 | 2mg 5mg |
SCH772984 有效抑制 ERK1 和 ERK2 活性。