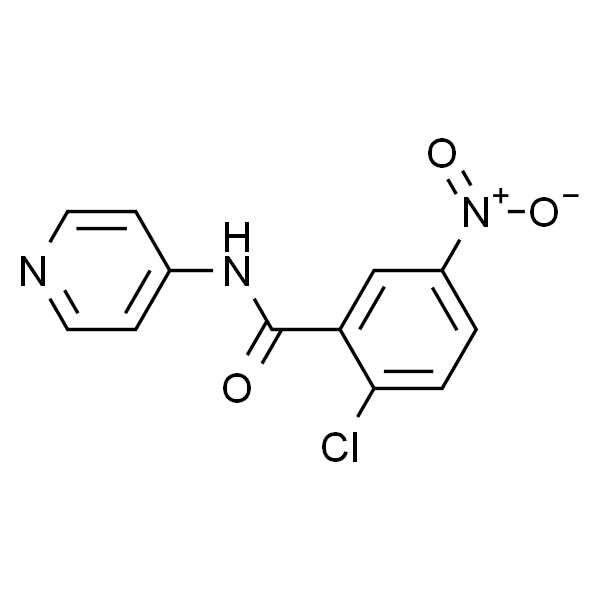
GSK-0660
货号:
IG2510
品牌:
Jinpan
暂无详情
产品简介
| 有效期 | 2年 |
| 描述 | 是有效的 PPARβ 和 PPARδ 拮抗剂。 |
| MDL | MFCD12828770 |
| 英文名称 | GSK-0660 |
| CAS | 1014691-61-2 |
| 分子式 | C19H18N2O5S2 |
| 分子量 | 418.49 |
| 储存条件 | 2-8℃ |
| 纯度 | ≥98% |
| 外观(性状) | Solid |
| 单位 | 瓶 |
| 生物活性 | GSK0660是有效的PPARβ/δ拮抗剂,pIC50为6.8。在浓度高达近于10 μM时,对PPARα和PPARγ没有活性。[1] |
| In Vitro | GSK0660 inhibits HRMEC (human retinal microvascular endothelial cells) proliferation and differentiation[2]. |
| In Vivo | GSK0660 is rapidly cleared and does not accumulate in the blood in vivo[1]. GSK0660 is efficacious against retinal NV when administered by IVIT or IP injection. Intravitreal injection has the advantages of producing high levels of drug at active sites of neovascular disease, but deleterious side effects are associated with this route of drug administration, including endophthalmitis, cataractogenesis, and glaucoma. Systemic administration could avoid these side effects, but it is hampered by the need for repeated dosing to obtain target concentrations of active drug in diseased tissues. It also needlessly exposes disease-free organs and tissues to active drug[2]. |
| SMILES | O=C(C1=C(S(=O)(NC2=CC=C(NC3=CC=CC=C3)C=C2OC)=O)C=CS1)OC |
| 靶点 | PPAR |
| 动物实验 | HRMECs were seeded in six-well plates at 2 × 105 cells/well and maintained under standard tissue culture conditions. At 80% confluency, the cells were serum starved for 12 hours, then treated on a background of 0.5% serum-containing vehicle (0.1% DMSO) or PPAR-β/δ agonist GW0742 (0.01, 0.1, or 1.0 μM) or on a background of 2% serum-containing vehicle or PPAR-β/δ antagonist GSK0660 (0.01, 0.1, or 1.0 μM) for 6 hours. Cells were washed twice with cold PBS and total RNA was collected. Total RNA isolated from the culture wells was reverse transcribed. Quantitative RT-PCR was performed.[2] |
| 细胞实验 | Animal Models: Sprague-Dawley rat; Dosages: 0.2 or 1.0 mg/kg; Administration: i.p.[2] |
| 数据来源文献 | [1] Shearer BG, et al. Mol Endocrinol. 2008, 22(2):523-9. [2] Megan E. Capozzi, et al. Invest Ophthalmol Vis Sci. 2013, 54( |
| 规格 | 2mg 5mg |