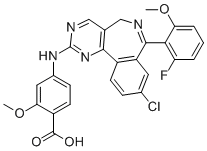
MLN8237CAS号: 1028486-01-2分子式: C27H20ClFN4O4分子量: 518.9描述纯度储存/保存方法别名可溶性/溶解性靶点In vitro(体外研究)In vivo(体内研究)
| 产品描述 | |
| 描述 |
MLN8237 (Alisertib) is a selective Aurora kinase A inhibitor with a median IC50 of 61 nM. |
| 纯度 |
>98%
|
| 储存/保存方法 |
Store at -20℃ for one year(Powder);Store at 2-4℃ for two weeks;Store at -20℃ for six months after dissolution.
|
| 基本信息 | |
| 别名 |
Alisertib;MLN 8237; MLN-8237
|
| 可溶性/溶解性 |
DMSO :27 mg/mL (52.03 mM)
|
| 生物活性 | |
| 靶点 |
Aurora A
|
| In vitro(体外研究) |
MLN8237 shows >200-fold higher selectivity for Aurora A than the structurally related Aurora B with an IC50 of 396.5 nM, and does not have any significant activity against 205 other kinases. MLN8237 (0.5 μM) treatment inhibits the phosphorylation of Aurora A in MM1.S and OPM1 cells, without affecting the Aurora B mediated histone H3 phosphorylation. MLN8237 significantly inhibits cell proliferation in multiple myeloma (MM) cell lines with IC50 values of 0.003-1.71 μM. MLN8237 displays more potent anti-proliferation activity against primary MM cells and MM cell lines in the presence of BM stroma cells, as well as IL-6 and IGF-1 than against MM cells alone. MLN8237 (0.5 μM) induces 2- to 6-fold increase in G2/M phase in primary MM cells and cell lines, as well as significant apoptosis and senescence, involving the up-regulation of p53, p21 and p27, as well as PARP, caspase 3, and caspase 9 cleavage. In addition, MLN8237 shows strong synergistic anti-MM effect with dexamethasone, as well as additive effect with doxorubicin and bortezomib. MLN8237 (0.5 μM) treatment causes the inhibition of colony formation of FLO-1, OE19, and OE33 esophageal adenocarinoma cell lines, and induces a significant increase in the percentage of polyploid cells, and subsequently an increase in the percentage of cells in the sub-G1 phase, which can be further enhanced in combination with cisplatin (2.5 μM), involving the higher induction of TAp73β, PUMA, NOXA, cleaved caspase-3, and cleaved PARP as compared with a single-agent treatment.
|
| In vivo(体内研究) |
MLN8237 significantly reduces the tumor burden with tumor growth inhibition (TGI) of 42% and 80% at 15 mg/kg and 30 mg/kg, respectively, and prolongs the survival of mice compared with the control.
|
分子结构图