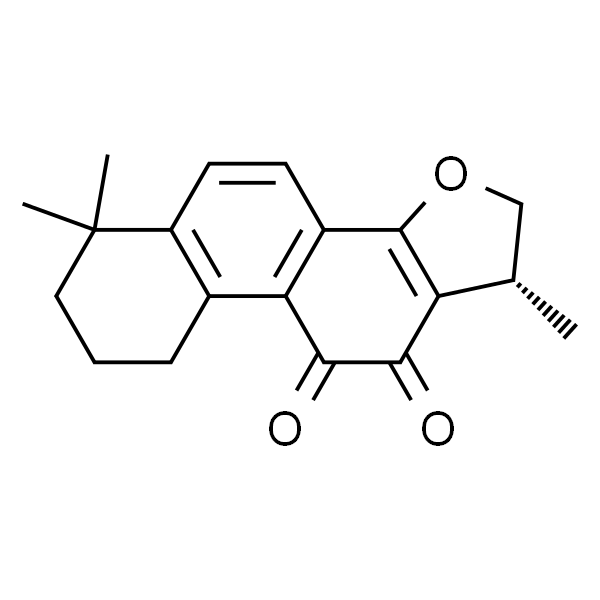
Cryptotanshinone 隐丹参酮
货号:
IC0590
品牌:
Jinpan
暂无详情
产品简介
| MDL | MFCD07636810 |
| 别名 | 隐丹参醌 |
| CAS | 35825-57-1 |
| 分子式 | C19H20O3 |
| 分子量 | 296.35 |
| 纯度 | HPLC≥98% |
| 单位 | 瓶 |
| 生物活性 | Cryptotanshinone是从丹参的根中提取的天然化合物, 具有抗肿瘤活性。 Cryptotanshinone抑制 STAT3 的 IC50 为4.6 μM[1-4]。 |
| IC50 | STAT3:4.6 μM (IC50) [1-4] |
| In Vitro | 与不显示活性的丹参酮IIA相比, 隐丹参酮显着抑制STAT3依赖性荧光素酶活性, STAT3 Tyr705磷酸化和STAT3的二聚化。隐丹参酮 (7μM) 显着阻断DU145细胞中STAT3 Tyr705磷酸化但不抑制STAT3 Ser727磷酸化, 并且显着抑制JAK2磷酸化, IC50为5μM, 而不影响上游激酶c-Src和EGFR的磷酸化, 表明抑制STAT3 Tyr705磷酸化可能是由于直接机制可能通过绑定到STAT3的SH2域。隐丹参酮通过阻断STAT3活性显着抑制具有组成型活性STAT3且GI50为7μM的DU145前列腺癌细胞的增殖, 其导致细胞周期蛋白D1, Bcl-xL和存活蛋白的下调, 随后在G0-G1中积累。相。隐丹参酮对PC3, LNCaP和MDA-MB-468细胞的生长抑制作用较小[1]。隐丹参酮显着减弱了DEX对卵巢的体外激素作用, 如T的显着降低和培养基中P水平的增加所示。隐丹参酮可显着提高DEX处理卵巢中磷酸化AKT2和GSK3β的水平[2]。伊马替尼和隐丹参酮的联合治疗在伊马替尼敏感和耐药CML细胞系以及原发性CML细胞中均显示出显着的协同杀伤作用[3]。 |
| In Vivo | Cryptotanshinone逆转卵巢IR并显着增加来自DEX处理的小鼠的所有检查组织中的2-脱氧-D- [1, 2-3H] – 葡萄糖摄取。隐丹参酮可显着降低排卵率和血浆E2和P水平[2]。隐丹参酮给药以剂量依赖性方式显着降低ob / ob小鼠 (C57BL / 6J-Lepob) 和饮食诱导的肥胖 (DIO) 小鼠的体重和食物摄入。隐丹参酮在脂肪组织中引起明显较少的脂肪, 血清甘油三酯和胆固醇水平显着降低, 并且骨骼肌的AMPK活性比对照小鼠高2.5至3倍。以600mg / kg /天口服给予隐丹参酮可显着降低ob / ob小鼠 (C57BL / 6J-Lepob) , db / db小鼠 (C57BL / KsJ-Leprdb) 和ZDF大鼠的血糖水平, 3天, 并持续整个监测期[4]。 |
| 激酶实验 | 用具有STAT3结合元件的报告质粒瞬时转染HCT-116细胞以调节荧光素酶测定。用Cryptotanshinone处理细胞24小时, 浓度范围为0.2至50μM。处理后, 在20μL被动裂解缓冲液中收获细胞, 并通过Wallac Victor2上的Dual Luciferase Reporter Assay试剂盒评估荧光素酶活性。抑制荧光素酶活性50%的隐丹参酮的浓度代表IC 50值。 |
| SMILES | O=C(C1=C2C=CC3=C1CCCC3(C)C)C(C4=C2OC[C@@H]4C)=O |
| 靶点 | STAT |
| 细胞实验 | 如前所述, MTT测定用于评估细胞生长抑制。将细胞以每孔8000个细胞的密度接种在含有10%FBS的RPMI-1640的96孔板中。加入不同浓度的伊马替尼和CPT并再孵育24小时。然后, 向每个孔中加入20μLMTT, 并在酶联免疫吸附测定 (ELISA) 读板仪上测量570nm处的吸光度。根据先前的研究确定伊马替尼和CPT之间的药物相互作用系数 (CDI) 。计算方法如下:CDI / AB / (A×B) 。根据各组的吸光度, AB为组合组与对照组的比例; A或B是单一药剂组与对照组的比率。 CDI 1表示拮抗作用。在组合治疗组中, CPT的浓度根据50%抑制浓度 (IC50) 值任意指定。然后, 在用不同浓度的伊马替尼加CPT (一种细胞类型的恒定CPT浓度) 处理后测定细胞活力。最后, 计算伊马替尼的组合IC 50值, 并在表I中表示。原发性CML细胞CP1至CP3分离自慢性期患者, 而BC1和BC2分离自急变期患者。进行三组独立的实验。 IC50值表示为平均值±标准偏差 (SD) 。 |
| 数据来源文献 | [1]. Shin DS, et al. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res. 2009 Jan 1; 69 (1) :193-202.
[2]. Huang Y, et al. Cryptotanshinone reverses ovarian insulin resistance in mice through activation of insulin signaling and the regulation of glucose transporters and hormone synthesizing enzymes. Fertil Steril. 2014 Aug; 102 (2) :589-596.e4. [3]. Ge Y, et al. Cryptotanshinone acts synergistically with imatinib to induce apoptosis of human chronic myeloid leukemia cells. Leuk Lymphoma. 2014 Jun 25:1-9. [4]. Kim EJ, et al. Antidiabetes and antiobesity effect of cryptotanshinone via activation of AMP-activated protein kinase. Mol Pharmacol. 2007 Jul; 72 (1) :62-72. Epub 2007 Apr 11. |
| 备注 | 以上数据均来自公开文献, Jinpan暂未进行独立验证, 仅供参考。These protocols are for reference only. Jinpan does not independently validate these methods. |
| 规格 | 10mg 10mM*1mL (in DMSO) 50mg |
可以抑制 STAT3 。具有抗肿瘤活性。