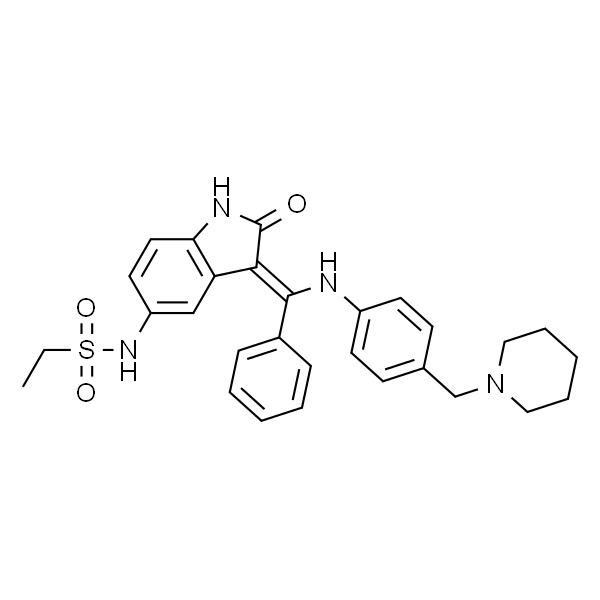
Hesperadin
货号:
IH0630
品牌:
Jinpan
暂无详情
产品简介
| MDL | MFCD18074526 |
| EC | EINECS 200-258-5 |
| 别名 | N-[(3Z)-2-氧代-3-[苯基-[4-(哌啶-1-甲基)苯胺]亚甲基]-1H-吲哚-5-基]乙烷磺酰胺 |
| 英文名称 | Hesperadin |
| CAS | 422513-13-1 |
| 分子式 | C29H32N4O3S |
| 分子量 | 516.65 |
| 纯度 | ≥98% |
| 单位 | 瓶 |
| 生物活性 | Hesperadin是ATP竞争型的Aurora B 抑制剂,IC50值为250 nM。[1-3] |
| IC50 | Aurora B:250 nM (IC50)[1-3] |
| In Vitro | Hesperadin抑制多种人类临床分离的甲型和乙型流感病毒,具有单一至亚微摩尔的功效,包括奥司他韦耐药菌株。机理研究表明,hesperadin通过延迟病毒核糖核蛋白复合物的核进入来抑制病毒复制的早期阶段,从而抑制病毒RNA转录和翻译以及病毒蛋白合成[2]。Hesperadin还以1μM药物浓度抑制其他激酶如AMPK,Lck,MKK1,MAPKAP-K1,CHK1和PHK。 Hesperadin在HeLa细胞中引起多倍性。 Hesperadin处理的HeLa细胞显示出对齐和分离缺陷,但姐妹染色单体分离是完整的。 Hesperadin导致有丝分裂和胞质分裂缺陷。 Hesperadin抑制Aurora B.免疫荧光显微镜检查显示,Hesperadin处理的细胞,其中染色体向相反的两极伸展,即已进入后期,未能组装中心纺锤体并正确定位人中枢心轴亚基CYK-4和MKLP1 [1] ]。由于Aurora B活性降低引起的多种有丝分裂缺陷的出现以及从有丝分裂染色体的动粒中消除检查点蛋白(如hBUBR1和CENP-E),Hesperadin抑制细胞增殖[3]。 |
| 激酶实验 | 用含有5μg组蛋白H1,1μMATP,1μCi[32P] ATP和适当浓度的Hesperadin或DMSO的20μL激酶缓冲液中的10μL珠子在37℃进行激酶测定30分钟。加入SDS样品缓冲液,煮沸样品并通过SDS-PAGE分离。将凝胶干燥,通过PhosphorImager分析检测放射性信号[1]。 |
| SMILES | O=C1NC2=CC=C(C=C2/C1=C(NC3=CC=C(C=C3)CN4CCCCC4)C5=CC=CC=C5)NS(CC)(=O)=O |
| 靶点 | Aurora Kinase |
| 细胞实验 | 为了确定Hesperadin的细胞毒性,向每个孔中加入200μL含有连续半对数稀释的Hesperadin的新鲜DMEM(不含FBS)培养基。在37℃下在细胞培养箱中用5%CO 2温育48小时后,除去培养基并加入含有40μg/ mL中性红的100μLDMEM培养基。将溶液在37℃下在细胞培养箱中再孵育4小时。除去培养基,通过加入100μL脱色溶液(50%乙醇,49%H 2 O和1%乙酸)溶解活细胞摄取的中性红的量。测定溶液在540nm处的吸光度[2]。 |
| 数据来源文献 | [1]. Hauf S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003 Apr 28;161(2):281-94. [2]. Hu Y, et al. Chemical Genomics Approach Leads to the Identification of Hesperadin, an Aurora B Kinase Inhibitor, as a Broad-Spectrum Influenza Antiviral. Int J Mol Sci. 2017 Sep 8;18(9). [3]. Ladygina NG, et al. Effect of the pharmacological agent hesperadin on breast and prostate tumor cultured cells. Biomed Khim. 2005 Mar-Apr;51(2):170-6. |
| 规格 | 5mg 10mg 50mg |
Hesperadin是ATP竞争型的Aurora B 抑制剂。