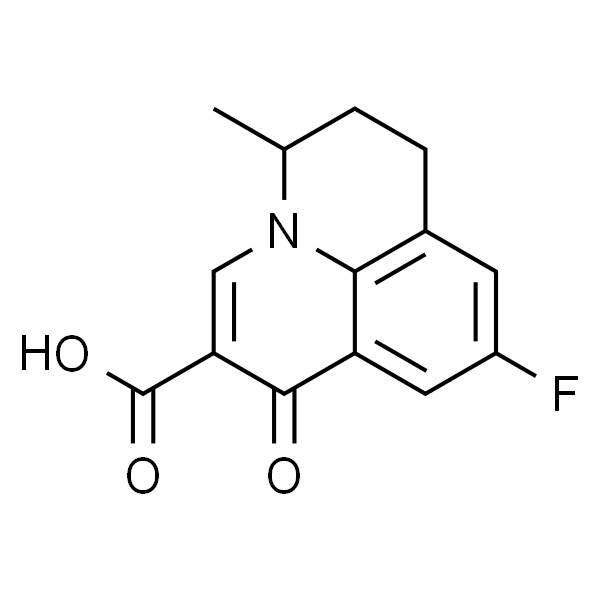
Flumequine 氟甲喹
货号:
IF0140
品牌:
Jinpan
暂无详情
产品简介
| MDL | MFCD00079298 |
| EC | EINECS 255-962-6 |
| 别名 | Apurone;Quinolone |
| CAS | 42835-25-6 |
| 分子式 | C14H12FNO3 |
| 分子量 | 261.25 |
| 纯度 | HPLC≥98% |
| 单位 | 瓶 |
| 生物活性 | Flumequine 是一种喹诺酮类抗生素,为Topoisomerase II抑制剂,IC50 值为 15 μM (3.92 μg/mL)[1-3]。 |
| IC50 | Topoisomerase II:15 μM [1-3] |
| In Vitro | Flumequine是拓扑异构酶II抑制剂,IC50为3.92μg/ mL,有效抑制Gyrase,IC50为1764μg/ mL。 Flumequine(0-625μg/ mL)可增加CHL细胞核DNA的迁移[1]。 Flumequine抑制西班牙猪痢疾短螺旋体分离株,MIC50和MIC90分别为50和100μg/ mL,MBC50和MBC90分别为50,200μg/ mL [2]。 Flumequine抑制A. salmonicida分离株,MIC范围为0.06至32μg/ mL [3]。 |
| In Vivo | Flumequine(0-500 mg/kg,po)在小鼠的胃,结肠和膀胱中引起剂量相关的DNA损伤,给药后1小时和3小时但不是24小时[1]。 |
| 激酶实验 | 将含有0.4μg运动前体DNA和1U拓扑异构酶II的20μL反应混合物和每种抗菌剂(包括Flumequine)的连续稀释液在37℃下在含有20mM Tris-HCl(pH7.5)的缓冲液中温育5分钟, 120mM KCl,10mM MgCl 2,1mM ATP,0.5mM二硫苏糖醇和30μg/ mL牛血清白蛋白。通过加入具有凝胶上样缓冲液的肌氨酸终止反应后,通过琼脂糖凝胶电泳分离链接和连接的kDNA。凝胶用溴化乙锭染色,并使用UV光(302nm)和凝胶印刷200i/VGA拍照,并用图像分析仪追踪条带的亮度。对每个条带进行定量,测量每种浓度的喹诺酮处理的DNA量,以确定对DNA促旋酶和拓扑异构酶II的50%抑制浓度[1]。 |
| SMILES | O=C(C1=CN2C(C)CCC3=C2C(C1=O)=CC(F)=C3)O |
| 靶点 | Bacterial;Topoisomerase |
| 动物实验 | 分别在4周和7周龄的婴儿和年轻成年雄性ddY小鼠在适应环境1周后使用。用氟尿喹以<500mg> |
| 细胞实验 | 中国仓鼠肺细胞系CHL/IU常规维持在单层培养物中,在Dulbecco改良的MEM培养基中,在37℃,5%CO 2气氛下补充有10%胎牛血清。用溶于DMSO中的Flumequine处理指数生长的细胞1小时。选择剂量范围是为了获得受损和高度受损的细胞。氟喹处理后,将细胞以1%溶解于盐水中的GP42琼脂糖中。确定每种剂量的细胞数和细胞活力[1]。 |
| 数据来源文献 | [1]. Kashida Y, et al. Mechanistic study on flumequine hepatocarcinogenicity focusing on DNA damage in mice. Toxicol Sci. 2002 Oct;69(2):317-21. [2]. Aller-Morán LM, et al. Evaluation of the in vitro activity of flumequine against field isolates of Brachyspira hyodysenteriae. Res Vet Sci. 2015 Dec;103:51-3. [3]. Giraud E, et al. Mechanisms of quinolone resistance and clonal relationship among Aeromonas salmonicida strains isolated from reared fish with furunculosis. J Med Microbiol. 2004 Sep;53(Pt 9):895-901. |
| 规格 | 10mg 10mM*1mL (in DMSO) 50mg 100mg |
是一种喹诺酮类抗生素,为拓扑异构酶 II (Topoisomerase II) 抑制剂。