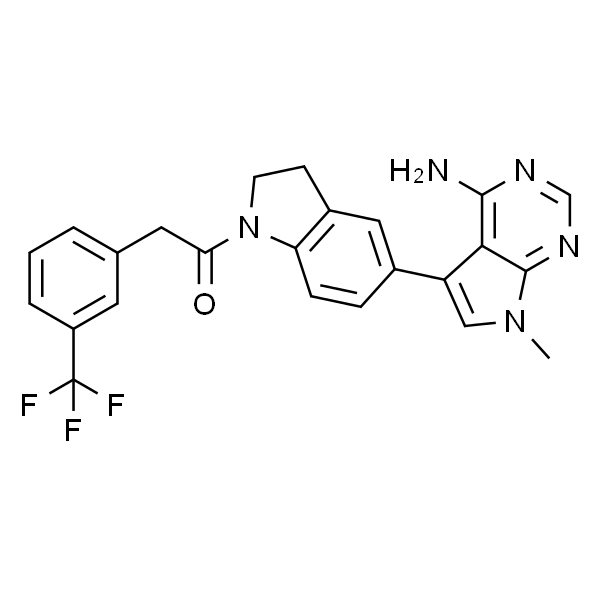
IC-87114
货号:
II0740
品牌:
Jinpan
暂无详情
产品简介
| 别名 | 2-[(6-Amino-9H-purin-9-yl)methyl]-5-methyl-3-(2-methylphenyl)-4(3H)-quinazolinone |
| 英文名称 | IC-87114 |
| CAS | 371242-69-2 |
| 分子式 | C22H19N7O |
| 分子量 | 397.43 |
| 纯度 | ≥98% |
| 单位 | 瓶 |
| 生物活性 | IC-87114 是一种有效的选择性 PI3Kδ 抑制剂,IC50 为 0.5 μM。[1-3] |
| IC50 | PI3Kδ:0.5 μM ; PI3Kγ:29 μM ; PI3Kβ:75 μM ; [1-3] |
| In Vitro | IC-87114(IC87114),原始抑制剂的类似物,并测试相对于其他I类PI3K的PI3Kδ选择性。 IC87114对PI3Kδ抑制的IC50为0.5μM,而PI3Kα,PI3Kβ和PI3Kγ的IC50值分别> 100,75和29μM。因此,相对于PI3Kγ,IC87114对PI3Kδ的选择性高58倍,相对于PI3Kα和PI3Kβ,选择性超过100倍。 IC87114在至少0.3-10μM的浓度范围内选择性地拮抗PI3Kδ[1]。 IC-87114(10μM)也用于选择性抑制PI3Kδ催化活性以解决该问题。 IC87114(10μM)在处理1小时后有效灭活巨噬细胞中的Akt(n = 6;与对照相比P <0.001)。 IC-87114(IC87114)的作用是下一次检测到的AP-1 DNA结合活性。电泳迁移率变动分析表明,用TNF-α(10 ng / mL; P <0.001)和TNF-α(20 ng / mL; P <0.001)处理后,AP-1的DNA结合活性显著增加。 。 IC87114在处理1小时后单独诱导AP-1 DNA结合活性。此外,IC87114(10μM)和TNF-α(0-20 ng / mL)共刺激后的AP-1 DNA结合活性强于仅用TNF-α治疗(0-20 ng / mL; n = 5) ; P <0.01)。 IC87114(10μM)也可有效抑制巨噬细胞中p110δ催化活性(Akt磷酸化),有或无TNF-α处理24小时(n = 6; P <0.001)[2]。 |
| In Vivo | 用PD 89059(10mg/kg),IC-87114(0.3mg/kg)和BAY 11-7085(10mg/kg)治疗显著(P 0.05)。值得注意的是,观察到的PD 89059,IC-87114和BAY 11-7085引起的组织病理学气道重塑的减少与AG 1478和SU6656所见的减少相比效果较差[3]。 |
| SMILES | O=C1N(C(CN2C3=C(C(N)=NC=N3)N=C2)=NC4=CC=CC(C)=C14)C5=C(C)C=CC=C5 |
| 靶点 | PI3K |
| 动物实验 | 小鼠[3] BALB/c小鼠在第0天腹膜内注射10μg卵白蛋白(OVA)在0.2ml alu-Gel-S中免疫一次。十天后,小鼠鼻内(in)用OVA(30μg)攻击在50μLPBS)或PBS中,每天一次,连续四天。为了研究ERK1/2,PI3Kδ和NF-κB是否是EGFR反式激活下游的信号传导效应子,建立了六个治疗组(AF,每组10-30只动物)。 A组和B组中的小鼠鼻内用0.2mL药物载体预处理。 C,D和E组分别以相同体积的三种不同药物(分别为PD 98059,IC-87114和BAY 11-7085)以10 mg/kg,10 mg/kg和0.3 mg/kg进行预处理。 F用地塞米松(1mg/kg),每次用OVA攻击前1小时。这些剂量选自以前的研究,它们被证明是有效的。 |
| 细胞实验 | 来自两种类型小鼠的鼠巨噬细胞细胞系RAW264.7和腹膜巨噬细胞维持在含有10%胎牛血清(FCS)的Dulbecco改良的Eagle培养基(DMEM)中。将培养物保持在37℃,在95%O 2加5%CO 2气氛的潮湿培养箱中。用不同浓度的TNF-α处理细胞并使用IC-87114(IC87114)在早期时间点(30分钟)TNF-α刺激前抑制PtdIns(3,4,5)P3依赖的Akt磷酸化[2] 。 |
| 数据来源文献 | [1]. Sadhu C, et al. Essential role of phosphoinositide 3-kinase delta in neutrophil directional movement. J Immunol. 2003 Mar 1;170(5):2647-54. [2]. Zheng L, et al. Inactivation of PI3Kδ induces vascular injury and promotes aneurysm development by upregulating the AP-1/MMP-12 pathway in macrophages. Arterioscler Thromb Vasc Biol. 2015 Feb;35(2):368-77. [3]. El-Hashim AZ, et al. Src-dependent EGFR transactivation regulates lung inflammation via downstream signaling involving ERK1/2, PI3Kδ/Akt and NFκB induction in a murine asthma model. Sci Rep. 2017 Aug 30;7(1):9919. |
| 规格 | 5mg 10mg 50mg 100mg |
IC-87114 是一种有效的选择性 PI3Kδ 抑制剂。