Isoimperatorin;异欧前胡素
货号:
II0370
品牌:
Jinpan
暂无详情
产品简介
| MDL | MFCD00272155 |
| EC | EINECS 801-715-8 |
| 别名 | 白芷甲素 |
| 英文名称 | Isoimperatorin |
| CAS | 482-45-1 |
| 分子式 | C16H14O4 |
| 分子量 | 270.28 |
| 纯度 | HPLC≥98% |
| 单位 | 瓶 |
| 生物活性 | Isoimperatorin 是Angelica dahurica 根的甲醇提取物, 有效抑制乙酰胆碱酯酶 (AChE) , IC50 为 74.6 μM[1]。 |
| IC50 | IC50: 74.6 μM (AChE) [1] |
| In Vitro | 在针对中草药新农药的筛选计划中, 发现羌活根茎的乙醇提取物对两种线虫, 松材线虫和南方根结线虫具有强烈的杀线虫活性。基于生物活性引导的分级分离, 从乙醇提取物中分离出四种成分, 并鉴定为Columbianetin, Falcarindiol, Falcarinol和Isoimperatorin。异欧前胡素对B. xylophilus的LC50值为21.83μg/ mL。当使用15分钟的紫外线光照处理时, 镰刀菌醇, 镰刀菌素和异欧前胡素对南方根结线虫的毒性几乎是黑暗处理的5倍, 而哥伦比亚的毒性仅为毒性的2倍。已经证明异欧前胡素具有对几种昆虫的杀虫活性, 如卷心菜蚜虫 (Brevicoryne brassicae) [2]。在白芷 (AD) 提取物的活性部分中鉴定出异欧前胡素[3]。异欧前胡素通常用作内标 (IS) [4]。 |
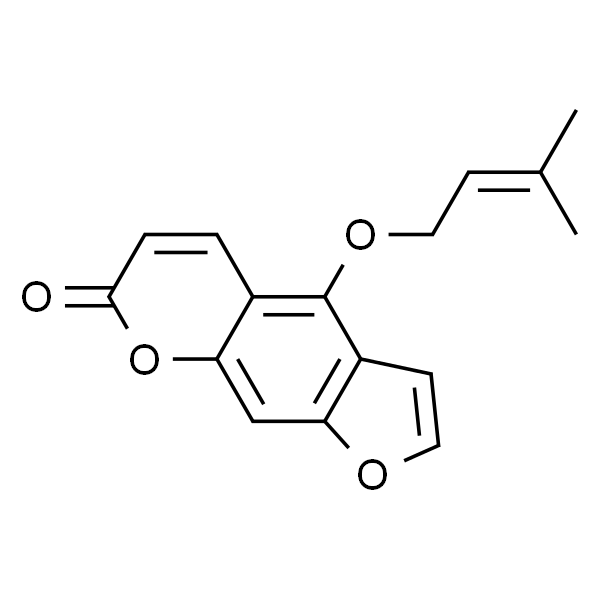
| SMILES | O=C1C=CC2=C(OC/C=C(C)/C)C3=C(OC=C3)C=C2O1 |
| 靶点 | AChE |
| 数据来源文献 | [1]. Kim DK, et al. Acetylcholinesterase inhibitors from the roots of Angelica dahurica. Arch Pharm Res. 2002 Dec; 25 (6) :856-9.
[2]. Liu G, et al. Identification of Nematicidal Constituents of Notopterygium incisum Rhizomes against Bursaphelenchus xylophilus and Meloidogyne incognita. Molecules. 2016 Sep 23; 21 (10) . pii: E1276. [3]. Park EY, et al. Angelica dahurica Extracts Improve Glucose Tolerance through the Activation of GPR119. PLoS One. 2016 Jul 8; 11 (7) :e0158796. [4]. Yu XA, et al. The pharmacokinetics, bioavailability and excretion of bergapten after oral and intravenous administration in rats using high performance liquid chromatography with fluorescence detection. Chem Cent J. 2016 Oct 14; 10:62. |
| 备注 | 以上数据均来自公开文献, Jinpan暂未进行独立验证, 仅供参考。 These protocols are for reference only. Jinpan does not independently validate these methods. |
| 规格 | 10mg 10mM*1mL in DMSO 20mg |
Isoimperatorin 可以有效抑制乙酰胆碱酯酶 (AChE)。