CUDC-101
货号:
IC1830
品牌:
Jinpan
暂无详情
产品简介
| 含量 | Purity≥98% |
| MDL | MFCD15528940 |
| 别名 | CUDC101 |
| CAS | 1012054-59-9 |
| 分子式 | C24H26N4O4 |
| 分子量 | 434.49 |
| 纯度 | ≥98% |
| 单位 | 支 |
| 生物活性 | CUDC-101 是一种高效的 HDAC、EGFR 和 HER2 抑制剂,对应的 IC50 值分别为 4.4、2.4 和 15.7 nM[1-4]。 |
| In Vitro | CUDC-101抑制I类和II类HDAC,但不抑制III类,Sir型HDAC。 CUDC-101在许多人类癌细胞类型中显示出广泛的抗增殖活性。 CUDC-101是一种有效的选择性HDAC,EGFR和HER2抑制剂,对下列蛋白激酶的抑制作用很弱(IC50):KDR(VEGFR2)(849 nM),Src(11000 nM),Lyn(840 nM),Lck (5910nM),Abl-1(2890nM),FGFR-2(3430nM),Flt-3(1500nM)和Ret(3200nM)[1]。 CUDC-101(300 nM)抑制全长AR(flAR)和AR变体AR-V7 [2]。 CUDC-101是筛选EGFR和HDAC抑制剂的所有三种ATC细胞系中最活跃的药物,8505c的最大抑制浓度(IC50)为0.15μM,C-643和SW-1736细胞的最大抑制浓度(IC50)为1.66μM 。 CUDC-101抑制癌细胞迁移并调节ATC细胞中的上皮 – 间质转化标志物表达。 CUDC-101还抑制HDAC和MAPK通路,诱导p21,并降低ATC细胞中survivin和XIAP的表达[3]。在处理的癌细胞中,CUDC-101(1μM)增加p53和α-微管蛋白(HDAC的非组蛋白底物)的乙酰化。 CUDC-101调节RTK活性和表达,并显示出对RTK和下游Akt信号传导的立即和稳定抑制[4]。 |
| In Vivo | CUDC-101(120mg/kg,iv,每日)在Hep-G2肝癌模型中诱导肿瘤消退,并且在其最大耐受剂量(MTD)下比厄洛替尼更有效。在厄洛替尼抗性A549 NSCLC异种移植模型中,CUDC-101(120mg/kg)显示出对肿瘤生长的有效抑制。在厄洛替尼敏感的H358 NSCLC模型中,CUDC-101(15,30,60mg/kg,iv)以剂量依赖性方式抑制肿瘤生长。 CUDC-101(120 mg/kg)在拉帕替尼耐药,HER2阴性,EGFR过度表达的MDA-MB-468乳腺癌模型和EGFR过度表达的CAL-27头颈部鳞状细胞癌(HNSCC)中引起显著的肿瘤消退。 )模型。 CUDC-101(120 mg/kg)也抑制K-ras突变体HCT116结肠直肠和表达EGFR/HER2(neu)的HPAC胰腺癌模型中的肿瘤生长[1]。在转移性ATC的体内小鼠模型中,CUDC-101抑制肿瘤生长和转移,并显著延长存活[3]。 CUDC-101(120 mg/kg)对异种移植模型中广泛的肿瘤类型有效[4]。 |
| 激酶实验 | 使用Biomol Color de Lys系统评估I类和II类HDAC的活性。简而言之,HeLa细胞核提取物用作HDAC的来源。在比色人工基质的存在下,将不同浓度的药物添加到HeLa细胞核提取物中。在测定结束时加入显色剂,并在Wallac Victor II 1420酶标仪中以405nM测量酶活性。 |
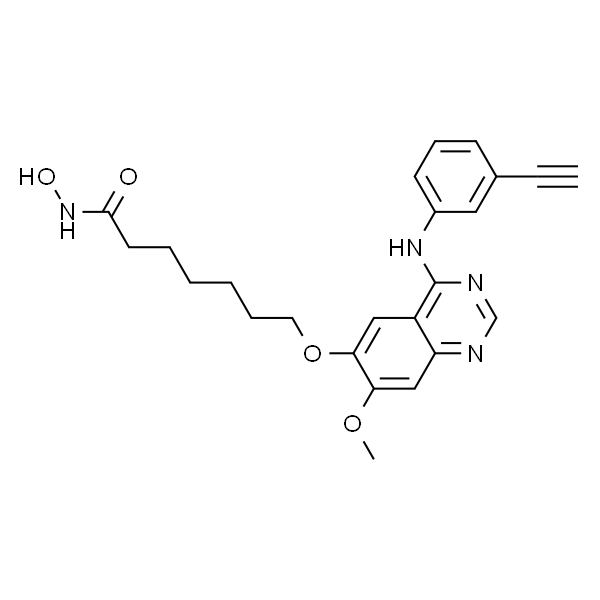
| SMILES | C#CC1=CC=CC(NC2=NC=NC3=CC(OC)=C(C=C23)OCCCCCCC(NO)=O)=C1 |
| 靶点 | EGFR;HDAC inhibitor |
| 动物实验 | 将4-6周龄雌性无胸腺小鼠(裸nu/nu CD-1)皮下接种到右后腹侧区域,在100-200μL的中等悬浮液中用1至5×10 6个细胞。对于乳腺癌细胞的原位植入,将100μL培养基中的细胞悬浮液通过27G针直接注射到乳腺脂肪垫中。如所示,口服,腹膜内或通过尾静脉注射施用不同剂量的CUDC-101,标准抗癌剂和媒介物。 |
| 细胞实验 | 将癌细胞系以每孔5000至10000个细胞接种在具有不同浓度化合物的96孔平底板中。在0.5%胎牛血清存在下,将细胞与化合物一起温育72小时。使用Perkin-Elmer ATPlite试剂盒通过三磷酸腺苷(ATP)含量测定评估生长抑制。 |
| 数据来源文献 | [1]. Xiong Cai et al Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer J. Med. Chem., 2010, 53 (5), pp 2000–2009 [2]. Sun H, et al. CUDC-101, a Novel Inhibitor of Full-Length Androgen Receptor (flAR) and Androgen Receptor Variant 7 (AR-V7) Activity: Mechanism of Action and In Vivo Efficacy. Horm Cancer. 2016 Jun;7(3):196-210. [3]. Zhang L, et al. Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget. 2015 Apr 20;6(11):9073-85. [4]. Lai CJ, et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity.Cancer Res. 2010 May 1;70(9):3647-56. Epub 2010 Apr 13 |
| 规格 | 5mg 10mg |
CUDC-101 是一种高效的 HDAC、EGFR 和 HER2 抑制剂。