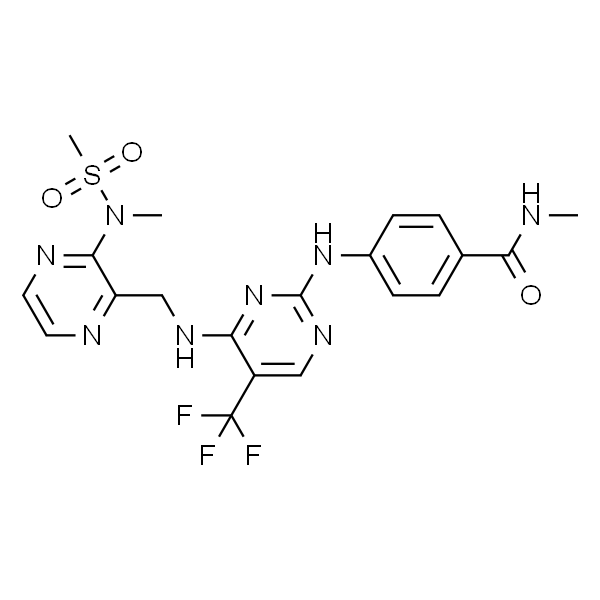
PND-1186
货号:
IP1870
品牌:
Jinpan
暂无详情
产品简介
| MDL | MFCD28125506 |
| 别名 | VS-4718; SR-2516 |
| CAS | 1061353-68-1 |
| 分子式 | C25H26F3N5O3 |
| 分子量 | 501.5 |
| 储存条件 | 2-8°C |
| 纯度 | ≥98% |
| 单位 | 瓶 |
| 生物活性 | PND-1186 是有效可逆的 FAK 抑制剂,细胞试验中的 IC50 为1.5 nM。[1-2] |
| IC50 | 1.5 nM (FAK)[1] |
| In Vitro | 在体外激酶测定中使用重组FAK激酶结构域作为谷胱甘肽-S-转移酶(GST)融合蛋白,PND-1186抑制FAK活性,IC50为1.5nM。通过对FAK Tyr-397的抗磷酸特异性免疫印迹测定,PND-1186在乳腺癌细胞中具有~100nM的IC 50。而1.0μMPND-1186(比IC50高5倍)对细胞增殖的影响有限,在非贴壁条件下或在软琼脂中作为球状体或集落生长时,0.1μMPND-1186阻断FAK和p130Cas酪氨酸磷酸化,促进caspase-3激活,并引发细胞凋亡。 PND-1186抑制4T1乳腺癌皮下肿瘤生长与肿瘤细胞凋亡和caspase 3活化升高相关[1]。 |
| In Vivo | 100 mg / kg PND-1186治疗显著降低最终4T1肿瘤重量2倍(n = 8,p 0.05)。与载体处理的对照相比,30和100mg / kg施用PND-1186显著增加肿瘤TUNEL染色。由于在PND-1186处理的小鼠的肿瘤中也发现了升高的裂解的半胱天冬酶-3染色[1]。 PND-1186显示多指数衰减,静脉内注射后终末半衰期(t1 / 2)为1.72小时。 ip和po给药后,PND-1186迅速被吸收,终末半衰期(t1 / 2)为2.15至2.65小时,生物利用度(%F)为14.8至42.2%。 PND-1186在腹膜内与口服给药后的生物利用度更高[2]。 |
| 靶点 | FAK |
| 动物实验 | 小鼠[1]使用六至八周大的雌性C57BL6和BALB / c小鼠。对于皮下肿瘤生长,将100μLPBS中的1×106mCherry标记的4T1细胞注射到Balb / C小鼠的后胁腹中。 8天后,将具有等体积肿瘤的小鼠(使用游标卡尺测量并通过长度×宽度2/2确定)分组(每组n = 8)并且将PND-1186溶解在PBS中的聚乙二醇400(PEG400)中(1: 1)每12小时以30mg / kg或100mg / kg皮下注射(100μL)颈部区域。对照动物接受PEG400:PBS注射,并在第13天,使用Olympus OV100活体荧光分子成像系统原位成像肿瘤,切除肿瘤并称重,一半在OCT中冷冻,一半溶解在蛋白质裂解缓冲液中用于FAK磷酸化分析。 |
| 细胞实验 | 对于细胞生长分析,将贴壁或悬浮的细胞用PND-1186处理指定的时间,通过有限的胰蛋白酶处理作为单细胞悬浮液收集,用70%乙醇固定,通过离心收集并用PBS洗涤。将细胞沉淀重悬于300μL含有碘化丙啶(PI)(10μg/ mL),不含DNA酶的RNA酶(100μg/ mL)的PBS中,然后在37℃下搅拌孵育1小时。通过流式细胞术分析样品,并通过ModFit LT3.2软件进行细胞周期分析。通过PI染色检测作为细胞凋亡的量度的二倍体DNA含量。对于细胞凋亡分析,用PND-1186处理贴壁或悬浮的细胞并如上收集,染色藻红蛋白(PE) – 缀合的膜联蛋白V结合和7-氨基 – 放线菌素(7-AAD)反应性,并在1小时内通过流式细胞术。象限门基于细胞自发荧光(阴性)星形孢菌素处理(阳性)对照定位。计算细胞凋亡为膜联蛋白V阳性细胞的百分比。在软琼脂测定中,通过目视检查至少200个细胞来定量细胞凋亡,并且定义为膜起泡或细胞收缩的外观。通过免疫印迹法在蛋白质裂解物中出现裂解的半胱天冬酶-3抗体反应性也可检测到细胞凋亡[1]。 |
| 数据来源文献 | [1]. Tanjoni I, et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol Ther. 2010 May 15;9(10):764-77. [2]. Walsh C, et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol Ther. 2010 May 15;9(10):778-90. |
| 规格 | 5mg 10mg |
PND-1186 是一种可逆的选择性FAK抑制剂