逆转素
货号:
IR0840
品牌:
Jinpan
暂无详情
产品简介
| 有效期 | 2年 |
| MDL | MFCD09953189 |
| 英文名称 | Reversine |
| CAS | 656820-32-5 |
| 分子式 | C21H27N7O |
| 分子量 | 393.23 |
| 储存条件 | 2-8°C |
| 纯度 | ≥98% |
| 外观(性状) | Powder |
| 单位 | 瓶 |
| 生物活性 | Reversine 是一种ATP-竞争性的 Aurora kinase 抑制剂,作用于 Aurora A,Aurora B 和 Aurora C,IC50 分别为 400,500 和 400 nM。[1-2] |
| In Vitro | Reversine是一种新型Aurora激酶抑制剂,可抑制人急性髓系白血病细胞的集落形成。 Reversine是Aurora A和B的有效抑制剂,也是Aurora C激酶的抑制剂。极光A和B活性被80%抑制,极光激酶C被抑制55%,已经浓度为0.5μM,而对其他激酶没有观察到抑制或仅有适度的抑制。在第二轮实验中,Reversine的IC50在Aurora激酶A上测定为400nM,而Aurora激酶B和C IC50分别为500和400nM。在MEK1上测定的IC50>1.5μM,肌肉肌球蛋白(非肌肉肌球蛋白II的类似物)的IC50为350nM [1]。 |
| In Vivo | Reversine和阿司匹林的组合可以更有效地诱导细胞周期停滞和细胞凋亡。为了评估该组合的抗肿瘤效果,通过皮下注射建立异种移植裸鼠模型。接种宫颈癌细胞的小鼠在肿瘤接种后约16天损失了其初始体重的约10%。然而,在组合组中,肿瘤生长(肿瘤重量)减少并且小鼠存活时间更长[2]。 |
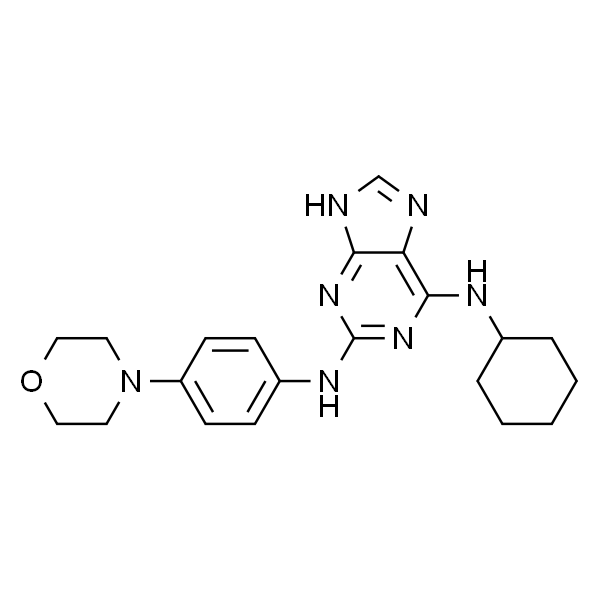
| SMILES | C1(NC2=CC=C(N3CCOCC3)C=C2)=NC(NC4CCCCC4)=C5C(NC=N5)=N1 |
| 靶点 | AuroraKinase |
| 动物实验 | 小鼠[2]使用雌性无胸腺6-8周龄BALB / c裸鼠。皮下注射U14细胞悬浮液(100μLRPMI-1640培养基中的5×10 6个细胞)。发展宫颈肿瘤的目的是产生用于药物治疗的组织学完整肿瘤。当肿瘤直径达到约1 cm时,每3天通过腹腔注射给予Reversine,阿司匹林或其组合,将这些小鼠中的25只随机分配到以下五组中的一组:(a)用小鼠治疗的小鼠RPMI-1640培养基,(b)用DMSO处理的小鼠,(c)用Reversine(10mg / kg)处理的小鼠,(d)用阿司匹林(1μg/ kg)处理的小鼠和(e)用Reversine处理的小鼠和阿司匹林组合。每周三次测量接种部位的体重和肿瘤大小。从两个直径每3天测量肿瘤大小,使用公式L×S2 / 2(L作为最长直径,S作为最短直径)估计肿瘤体积。 |
| 细胞实验 | 将HCT116和HL60细胞与5μmol/ L Reversine或0.01%DMSO一起温育。收获细胞并在70%乙醇中固定过夜。用PBS双洗后,用细胞周期染色试剂PBS,0.1%Triton X-100,200μg/ mL无DNA酶RNase和25μg/ mL碘化丙啶标记细胞,并在室温下在黑暗中孵育30分钟。 。使用FACSCalibur分析DNA含量。使用ATPlite 1step评估不同肿瘤细胞系的细胞活力。简言之,在新月体积的Reversine存在下,将每孔2×10 4个细胞接种在96孔板中。 72小时后,回收平板并向每个孔中加入100μLATPlite溶液。将板在700rpm下振荡2分钟并使用EnVision Multilabel读板仪测量发光。每个样品一式三份进行分析[1]。 |
| 数据来源文献 | [1]. D’Alise AM, et al. Reversine, a novel Aurora kinases inhibitor, inhibits colony formation of human acute myeloid leukemia cells. Mol Cancer Ther. 2008 May;7(5):1140-9. [2]. Qin HX, et al. Synergistic antitumor activity of reversine combined with aspirin in cervical carcinoma in vitro and in vivo. Cytotechnology. 2013 Aug;65(4):643-53. |
| 规格 | 5mg 10mg 25mg |
是一种ATP-竞争性的Aurorakinase抑制剂。