| 有效期 |
2年 |
| MDL |
MFCD00005240 |
| EC |
EINECS 215-925-7 |
| InChIKey |
XKVUYEYANWFIJX-UHFFFAOYSA-N |
| InChI |
InChI=1S/C4H6N2/c1-4-2-3-5-6-4/h2-3H,1H3,(H,5,6) |
| PubChem CID |
15073 |
| 别名 |
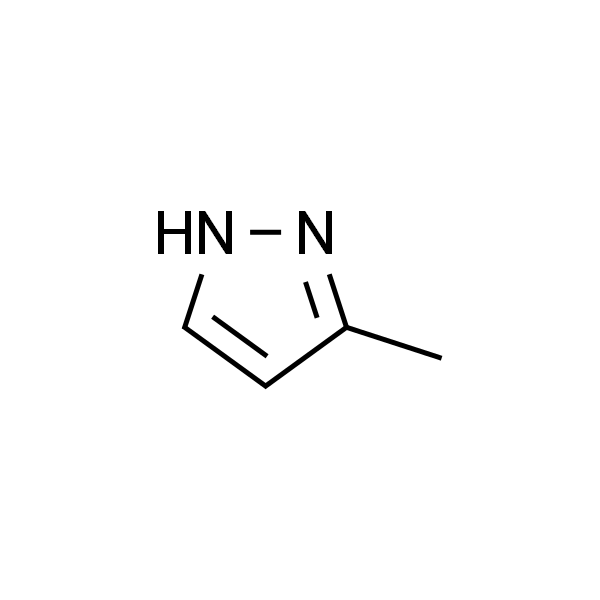
3-甲基-2H-吡唑 |
| 英文名称 |
3-Methylpyrazole |
| CAS |
1453-58-3 |
| 分子式 |
C4H6N2 |
| 分子量 |
82.1 |
| 储存条件 |
2-8℃ |
| 纯度 |
≥98% |
| 外观(性状) |
Light yellow to reddish brown Powder |
| 单位 |
瓶 |
| 生物活性 |
3-Methylpyrazole (3-MeP), a very weak inhibitor of ADH,could inhibit the metabolism of MAM by rat liver microsomes[1]. |
| In Vitro |
The addition of varying amounts of 3-MeP or 4-IP to liver microsomal incubations resulted in the inhibition of MAM metabolism. significant inhibition (approximately 20% with respect to control incubations without inhibitor) was obtained with a concentration of 4-IP of 0.001 raM. At the same concentration, little inhibition by 3-MeP was evident; however, a 25% inhibition of the metabolism resulted when the concentration of 3-MeP was in_x005f�creased to 0.01 mM.[1] |
| In Vivo |
Pretreatment of rats with either 4-IP or 3-MeP 15 min prior to treatment with 25 mg/kg MAMOAc produced a dose-dependent inhibition of liver DNA methylation, assayed 6 h after administration of the carcinogen. In the case of pretreatment with 3-MeP, the means of decreases in the levels of the methylated guanines were 25%, 31% (P < 0.05), and 49 % (P < 0.0 J), for respective do ses of 0.1, 0.3, and 1.0 mmol/kg of the inhibitor. In the colon mucosa, pretreatment with 3-MeP produced no inhibition of DNA methylation in the colon mucosa, rather an increase was observed. Thus, with pretreatments of 3-MeP of 0.1, 0.3, and 1.0 mmol/kg, the percent increases in colon mucosa levels of O6-MeG and 7-MEG were 37%, 56% (P< 0.03), and 52% (P < 0.01), respectively.[1] |
| 激酶实验 |
Rats were sacrificed by decapitation, and livers quickly excised, rinsed with ice-cold 0.9% NaC1 (saline), and homogenized in 3 weight volumes of ice-cold 0.25 M sucrose, 0.01 M potassium phosphate buffer, pH 7.5. The homogenate was centrifuged at 9000 g for 20 rain, and the microsomal fraction was obtained by centrifuging the resulting supernatant at 100000 g for 1 h. Microsomes were washed by resuspension in the same medium used for homogenization, followed by recentrifugation. The sediment from this step was weighed and resuspended in 3 weight-volumes of 0.1 M potassium phosphate buffer, pH 7.0. Following assay of protein content (Lowry et al. 1951), aliquots of resuspended microsomes (approximately 1 mg protein) were incubated, in a total volume of 0.5 ml, with 50 nmol MAM(1,2-14C), 3.5 mM glucose-6-phosphate, 5 units glucose-6- phosphate dehydrogenase, 1.5 mM NADP, 3.5 mM MgC12, and various concentrations of 3-MeP or 4-IP, in 0.1 M potassium phosphate buffer, pH 7.0. After incubation in a shaker bath at 37 ~ for 30 min, the reaction was stopped by plunging the incubation vessels into an ice bath. The suspensions were clarified and deproteinized by centrifugal ultrafiltration 600g, 30rain, 0-4~ using Centrifree Micropartition System tubes. Aliquots of the ultrafiltrates were submitted to HPLC using two Whatman ODS-3 (0.46 x 25 cm) columns in series, preceded by a 0.7 x 5 em column packed with Aminex A-29 anion exchange resin in the phosphate form. The system was eluted with 0.2 M ammonium phosphate buffer, pH 3.1, at a flow rate of 0.5 ml/min for the first 28 rain, and at 1 ml/min thereafter. The function of the Aminex A-29 precolumn was to retard the elution of formic acid (pK~ = 3.77) which would otherwise coelute with formaldehyde, very near the void volume. The absorbance of the effluent was monitored at 215 nm, and fractions were collected at 1 rain intervals for determination of radioactivity by scintillation counting. The extent of metabolism was determined by summing the radioactivity in the formaldehyde, methanol, and formic acid peaks, and subtracting from this the radioactivity found in the formaldehyde and methanol peaks which resulted when identical incubations were carried out in the absence of microsomes. Under these conditions, 2.4 nmol MAM was metabolized to prod_x005f�ucts in 30 min.[1] |
| SMILES |
CC1=NNC=C1 |
| 靶点 |
Others |
| 动物实验 |
Determination of methylated guanines in rat liver and colon mucosa DNA. Rats were injected i.p. with saline or with various concentra_x005f�tions of 3-MeP or 4-IP in saline. After 15 min, the animals were in�jected s.c. with 25 mg/kg of nonradioactive MAMOAc dissolved in saline. The rats were sacrificed 6 h later and the livers and colons rinsed with ice-cold 0.14 M KC1 and stored at -70 ~ DNA from livers and colon mucosae was isolated by the method of Daoud and Irving (1977). Purified DNA was hydrolyzed in 0.1 N HC1 at 37 ~ for 18 h. After determination of absorbance at 266 nm, portions of the hydrolyzates were submitted to HPLC using a Whatman Partisil SCX column (0.46 x 25 cm) with an Aquapore CX-300 guard column. The system was eluted with 0.1 M ammonium phosphate buffer, pH 2.0, at a rate of 2 ml/min. The effluent was sequentially monitored for absorbance at 276 nm for the quantitation of guanine, and for fluorescence at 370 nm (excitation, 295 nm) for the quantitation of O6-MeG and 7- MeG (Herron and Shank 1981).[1] |
| 数据来源文献 |
[1]. Fiala ES, et al. Differential effects of 4-iodopyrazole and 3-methylpyrazole on the metabolic activation of methylazoxymethanol to a DNA methylating species by rat liver and rat colon mucosa in vivo. J Cancer Res Clin Oncol. 1987;113(2):145-50. |
| 规格 |
200mg 500mg 1g |