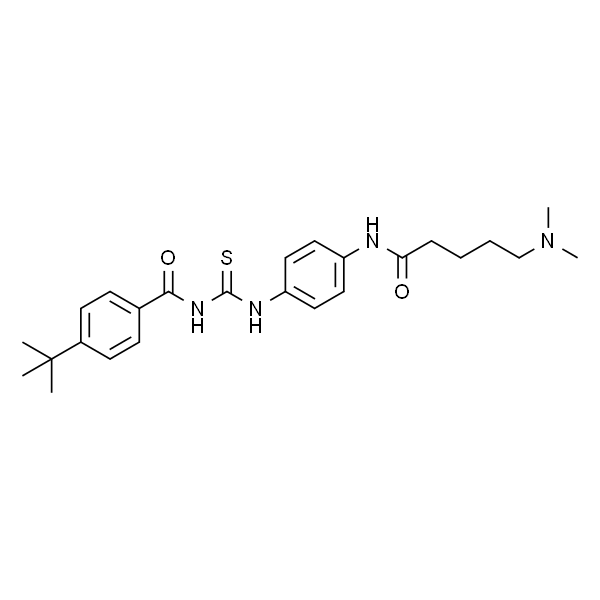
Tenovin-6
货号:
IT1590
品牌:
Jinpan
暂无详情
产品简介
| EC | EINECS 200-256-5 |
| 别名 | Tenovin6 |
| 英文名称 | Tenovin-6 |
| CAS | 1011557-82-6 |
| 分子式 | C25H34N4O2S |
| 分子量 | 454.63 |
| 储存条件 | 2-8°C |
| 纯度 | Purity≥98% |
| 单位 | 瓶 |
| 生物活性 | Tenovin-6 是水溶的 SIRT1、SIRT2 抑制剂,p53 的激活剂,同时能抑制 HDAC8 的活性,对 SirT1 peptide deacetylase,SirT2 和 SirT3 的 IC50 值分别为 21 μM,10 μM 和 67 μM。[1-4] |
| IC50 | SIRT2:10 μM ; SIRT1:21 μM ; SIRT3:67 μM [1-4] |
| In Vitro | Tenovin-6(0至15μM)剂量依赖性地增加不同细胞类型中LC3-II的水平,并且该增加是ATG5/7依赖性的。在存在或不存在Torin 1的情况下,Tenovin-6处理也增加了自噬囊泡的数量和强度,并且防止了Torin 1诱导的SQSTM1/p62降解。 Tenovin-6影响自溶酶体的酸化并损害溶酶体的水解活性,但不影响自噬体和溶酶体之间的融合。 tenovin-6抑制自噬与p53活化无关,敲除或敲除对SIRT1/2的抑制作用不能模拟tenovin-6对LC3B积累的影响[2]。 Tenovin-6抑制酿酒酵母培养物的生长,IC50为30μM,对酵母的毒性大于水溶性较低的tenovin-1。 Tenovin-6迅速增加MCF-7细胞中内源性K382-Ac p53的水平[1]。Tenovin-6(0,1,2.5,5或10μM)在所有OCI-Ly1,DHL-10,U2932,RIVA,HBL1和OCI-Ly10细胞系中以剂量和时间依赖性方式有效抑制细胞增殖。 Tenovin-6通过抑制经典自噬途径而不激活p53,一致地增加DLBCL细胞系中的LC3B-II水平,并且该增加不依赖于SIRT1/2/3和p53。 Tenovin-6通过外在细胞死亡途径诱导细胞凋亡[3]。 Tenovin-6抑制UM细胞的生长,对于92.1,Mel 270,Omm 1和Omm 2.3细胞,IC50分别为12.8μM,11.0μM,14.58μM和9.62μM[4]。 |
| In Vivo | Tenovin-6(50 mg/kg,ip)抑制小鼠肿瘤的生长[1]。 |
| 激酶实验 | 相关的FdL底物以7μM使用,NAD +以1mM使用。将Tenovins溶解在DMSO中,反应中的最终DSMO浓度小于0.25%。对于SirT1和HDAC8,每个反应使用一个酶单位,对于SirT2和SirT3,每个反应使用五个单位。反应在37℃下进行1小时。 |
| SMILES | O=C(NC(NC1=CC=C(NC(CCCCN(C)C)=O)C=C1)=S)C2=CC=C(C(C)(C)C)C=C2 |
| 靶点 | Sirtuin |
| 动物实验 | 雌性SCID小鼠皮下注射悬浮在基质胶中的1×106个ARN8细胞。允许肿瘤达到约10mm 3的尺寸。通过腹膜内注射每天以50mg/kg施用Tenovin-6。用含有环糊精20%(w/v)和DMSO 10%(v/v)的载体溶液处理对照动物。 |
| 细胞实验 | 将UM细胞接种到96孔板的每个孔中(5,000个细胞/孔),并且第二天用对照或Tenovin-6以0至20μM的递增浓度处理68小时,然后以20μL添加MTS。 /或者在波长为490nm处读取,通过S形剂量 – 响应曲线的曲线拟合确定IC50。 |
| 数据来源文献 | [1]. Lain S, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008 May;13(5):454-63. [2]. Yuan H, et al. Tenovin-6 impairs autophagy by inhibiting autophagic flux. Cell Death Dis. 2017 Feb 9;8(2):e2608. [3]. Yuan H, et al. Tenovin-6 inhibits proliferation and survival of diffuse large B-cell lymphoma cells by blocking autophagy. Oncotarget. 2017 Feb 28;8(9):14912-14924. [4]. Dai W, et al. Class III-specific HDAC inhibitor Tenovin-6 induces apoptosis, suppresses migration and eliminates cancer stem cells in uveal melanoma. Sci Rep. 2016 Mar 4;6:22622 |
| 规格 | 5mg |
Tenovin-6 是SIRT1、SIRT2 抑制剂,p53 的激活剂,同时能抑制 HDAC8 的活性。